

هیپوتونی شیرخواران:درمان ها
اسفند ۲۰، ۱۴۰۲
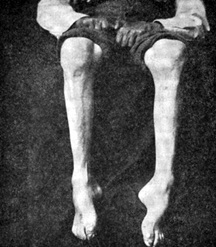
داروی تجاوز در مشروب
اسفند ۲۳، ۱۴۰۲
هیپوتونی شیرخواران:سطح آسیب
هیپوتونی مغزی
هیپوتونی مادرزادی خوش خیم ( Benign congenital hypotonia): این اختلال بطورعطف به ماسبق به نوزادانی اطلاق می شودکه دربدوتولدیااندکی پس ازآن ، هیپوتون بوده ولی بعدازآن ،تون آنهانرمال می شود.این اختلال شامل فرایندهای مختلفی است که مغز، واحد حرکتی ،یاهردوراگرفتارمی سازد.اکثراین نوزادان ، هیپوتونی مغزی دارند.علیرغم بهبودتون عضلانی ،این نوزادان درمعرض شیوع بالای عقب ماندگی مغزی ،مشکلات آموزشی ،وسایرعوارض ناهنجاری مغزی قراردارند.
اختلالات کروموزمی:صرف نظرازتنوع دراین اختلالات ، ویژگی مشترک اینگروه وجوددیسمورفیسم دستهاوصورت وهیپوتونی شدیداست.ازاین گروه می توان مواردزیررانام بر:تریزومی ها ،سندروم ترنر، سندروم کابوکی ،
| Trisomy 13, Patau syndrome
شکاف کام ، اغلب درمیانه ،فلکسیون انگشتان دست همراه با پلی داکتیلی ،هیپوتلوریسم ،بینی قلنبه ، گوش های بدشکل وباقرارپائین ، جمجمه کوچک وغیرعادی،ناهنجاری مغزی بخصوص هولوپروزنسفالی ، میکروفتالمی ،ناهنجاری قلبی ،نقائص اسکالپ ،هیپوپلازی یافقدان دنده ها ،آنومالی های احشاء وژنیتال |
 |
| Trisomy 18, Edwards syndrome
وزن تولد اندک ،مشت بسته باانگشت اشاره روی انگشت سوم و انگشت پنچم روی چهارم ،هیپ باریک باکاهش ابداکسیون ،استرنوم کوتاه ،پای چنبری ،میکروسفالی ، اکسی پوت برجسته ،میکروگناتی ،ناهنجاری قلبی وکلیوی ،وعقب ماندگی ذهنی |
 |
| Trisomy 21, Down syndrome
هیپوتونی ،صورت پهن ،فیسورهای پلکی روبه بالاواریب با چین اپی کانتال ،عنبیه منقوط ،درجات مختلفی ازعقب ماندگی ذهنی ورشدی، دیسپلازی لگن ،ناهنجاری قلبی ،شیارسیمیان ،دست های کوتاه وپهن ، هیپوپلازی فالانکس میانی انگشت پنجم ،آترزی روده،کام باقوس عمیق. |
 |
سندروم دوپلیکاسیون MECP2:این سندروم تنهادرپسرهادیده می شود،وراثت آن بصورت وابسته به X می باشد.ویژگی های آن ،عقب ماندگی شدیدمغزی ،فقدان صحبت کردن،اسپاستیسیته شدید،عفونتهای مکررریوی،وتشنج است.خصوصیات دیسمورفیک شامل براکی سفالی ،هیپوپلازی میانه صورت،گوشها ی بزرگ ،وپل بینی پهن است.
سندروم پرادرویلی : مشخصات آن شامل است بر:کاهش حرکات جنینی،دیسلوکاسیون هیپ ،کلاب فوت ،هیپوتونی ،کریپتورکیدیسم ،هیپوگنادیسم ،عقب ماندگی ذهنی ،قدکوتاه وچاقی.تقریبا 70% بیماران دچارحذ ف قطعه ای ازبازوی بلندکروموزوم 15(q11-13) پدری هستند.آنهائیکه دچاراین حذف نیستند،مشکل دیزومی مادری(هردوکروموزوم 15 ازمادراست) دارند.[دیزومی پدری درکروموزوم 15، منجربه سندروم آنجلمن می شود].البته هیپوتونی ومشکلات تغذیه ای تا8 الی 11 ماهگی ادامه داشته وسپس تون نرمال وگرسنگی مفرط جایگزین آنهامی شود. تشخیص براساس بررسی کروموزمی است.
| سندروم پرادرویلی | 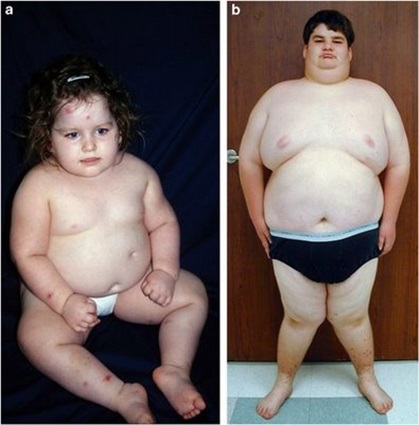 |
| ویژگی های خاص صورت دربیمارمبتلابه سندروم پرادرویلی دقت کنید. | 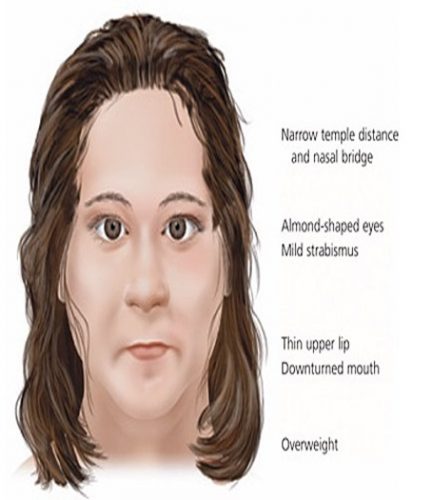
|
| سندروم کابوکی (Kabuki)
وراثت اتوزوم غالب یا وابسته به X می باشد.یافته هاشامل هیپوتونی ،مفاصل انعطاف پذیر،مشکلات تغذیه ای،مشکلات رفتاری ،عفونت مکرر، نقائص قلبی وکلیوی ،میکروگناتی ،وزن گیری ناگهانی طی بلوغ ومشکلات ایمنولوژیک می باشد. |
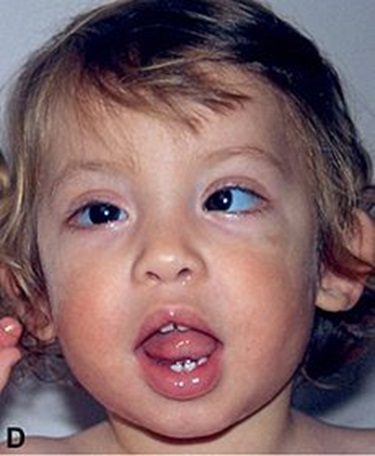 |
انسفالوپاتی مزمن غیرپیشرونده:دیسژنزی مغزی می تواندناشی ازعوامل مشخص یانامشخص محیطی ، اختلالات کروموزومی ،یا نقائص ژنتیک باشد.هیپوتونی معمولادربدوتولد وجودداشته وباگذشت زمان، بهترمی شود.زمانی که هیپوتونی توام باناهنجاری درسایراعضاء یا اندازه وشکل سرباشد ،بایدبفکردیسژنزی مغزی بود.تشخیص بیماری بکمک MRI میسر است.آسیب مغزی دردوران پری ناتال وگاهی درطی نوزادی بعلت آنوکسی ،هموراژ،عفونت وترومارخ می دهد.شروع ناگهانی هیپوتونی درنوزادی که قبلا نرمال بوده است ،صرف نظرازوجودیافقدان انسفالوپاتی،همیشه بیانگرعلل مغزی است.کاهش حرکات نوزادی می تواندبیانگرهموراژداخل بطنی باشد.هیپوتونی ، اولین ویژگی مننژیت است.رفلکس های تاندونی درطی فازحادممکن است کاهش یافته یانباشند.
اختلالات ژنتیک
دیس اتونومی فامیلیال ( Riley-Day Syndrome ):یک اختلال اتوزوم مغلوب شایع دریهوداشکنازی است.ویژگیهای شایع شامل است بر:آسپیراسیون مکونیال ،رفلکس ساکینگ ضعیف یافقدان آن ،وهیپوتونی.رفلکس های تاندونی هیپواکتیویا هیچ است.مشکل تغذیه ای معمولاوجودندارد.ساکینگ وبلع بطورجداگانه نرمال هستند،اماهماهنگ نیستند.سایریافته هاعبارتنداز:رنگ پریدگی،عدم ثبات حرارتی،فقدان پاپی قارچی زبان ،اسهال ،واتساع شکم.
سندروم اوکلوسربرورنال(Lowe Syndrome):ناشی ازکاهش فعالیت آنزیم اینوزیتول پلی فسفات 5 فسفاتاز OCRL1می باشد.انتقال وابسته به x می باشد.تظاهردرزنان ناقل بصورت کدورت قرنیه است.ویژگی های بالینی:شامل گرفتاری چشم(تمامی پسرهای مبتلادچارکاتاراکت[جدول 16.2] ونیمی دچارگلوکوم هستند)،مغز،وکلیه است.تکامل موتوری به آهستگی است وهمه پسرها اندکی مشکلات ذهنی دارند.مشکل کلیوی بصورت اختلال شبیه فانکونی است که باگذشت 10تا20 سال منجربه نارسائی کامل کلیه می شود.تشخیص براساس علائم بالینی وتائیدآن بکمک شناسائی کاهش فعالیت آنزیم درفیبروبلاستهای پوستی است.
| گلوکوم درسندروم لو |  |
| سندروم لو |  |
اختلالات پروکسی زومال:پروکسی زوم ، ارگانل هائی هستندکه دربیوسنتزفسفولیپیدهای اتری واسیدهای صفراوی ، اکسیداسیون اسیدهای چرب خیلی طولانی ،و کاتابولیسم فیتانات ها ، پیپه کولات، وگلیکولات دخالت دارند.جهش درژنهای مسئول ، منجربه این اختلالات می شود.طیف بالینی این اختلالات شامل نوع اولیه ، سندروم سربروهپاتورنال (Zellweger) ، ونیزآدرنولکودیستروفی نوزادی و بیماری رفسام (Refsum) نوزادی است.دومورداخیر،شدت کمتری دارند.ویژگی های بالینی:هیپوتونی شدید،آرتروگریپوزیس ،وخصوصیات دیسمورفیک. ضعف درساکینگ وگریه ،فقدان ویاضعیف بودن رفلکس های تاندونی.ناهنجاری کرانیوفاسیال(شامل سرگلابی شکل،سوتورهای پهن ،میکروگناتی ،کام بلند،پهن بودن پل بینی ،وهیپرتلوریسم)، ناهنجاری عضوی(سیروزصفراوی ،کلیه پلی کیستیک ،اختلالات مغزی )، تشنج نوزادی شایع است.تشخیص بابررسی بیوشیمیائی خون یاادرار، روی فیبروبلاست ها، می باشد.
کمبودپیرووات کربوکسیلاز:هیپوتونی ، تاکی پنه ،واختلالات حرکتی همراه باهیپوگلیسمی ،اسیدوزلاکتیک ،وهیپرسیترولینمی ازیافته های بارزاست.درمان:بارژیم غذائی بکمک تجویزتری هپتانوئین(triheptanoin ) وسیترات است.
سایرنقائص متابولیک:اختلالات مادرزادی متابولیسم می توانندباهیپوتونی تظاهرکنند.کمبوداسیدمالتاز وگانگلیوزیدوزژنرالیزه GM1 ازنمونه های آن است.
| دیسمورفیسم کلاسیک درسندروم زلوگر(تصویرچپ) و
شکل جمجمه درنوزاد مبتلا به سندروم زلوگر(تصویرراست) |
 |
 |
| rhizomelic chondrodysplasia punctata
به صورت دیسمورفیک ، ازجمله پیشانی برجسته ، هیپوپلازی میانه صورت ، فیلتروم بلند وحاشیه نازک ورمیلیون فوقانی توجه کنید.
|
 |
|
| آدرنولکودیستروفی |  |
|
اختلالات نخاعی
میلوپاتی هیپوکسیک- ایسکمیک:نوزادان مبتلابه آسفیکسی پری ناتال ،هیپوتون وفاقدرفلکس هستند.علت اصلی هیپوتونی،آسیب مغزی ولی گاهی نیزبعلت اختلال نخاعی است.نکروزایسکمیک ماده خاکستری بطورهمزمان درمغزونخاع دیده میشود.گرفتاری نخاعی بکمک بررسی EMG ، قابل تائیداست.
آسیب نخاعی:آسیب طناب نخاعی تنهادردوره نوزادی درتشخیص افتراقی هیپوتونی قرارمیگیرد.آسیب نخاع گردنی تقریبا همیشه درطی زایمان است(که حدود75% آن ناشی اززایمان بریچ و25% بعلت سفالیک است).کاهش سطح هوشیاری بعلت زایمان مشکل وطولانی ، دراین نوزادان شایع است وبنابراین هیپوتونی بطورکاذب به آسفیکسی یاترومای مغزی ارتباط داده می شود.اما وجوداختلال اسفنکتروکاهش حس زیرسطح میانه قفسه سینه بایداحتمال میلوپاتی رامطرح کند.
آسیبهای نمایش بریچ:آسیب ناشی ازکشش درناحیه تحتانی گردنی وفوقانی توراسیک تقریبادرمواردی که سرجنین بیش از90% اکستانسیون یابد،دیده می شود.ویژگیهای بالینی:هیپوتونی ،هموراژدرپوستریورفوسا( تظاهربصورت کاهش سطح هوشیاری ، آتونی ، کوادری پلاژی فلاسید ،وتنفس دیافراگماتیک دربدوتولد)، فقدان رفلکس های تاندونی وحرکات خودبخودی دراندام تحتانی ، فقدان تعریق زیرناحیه آسیب.تشخیص با MRI می باشد.
آسیب نمایش سفالیک:چرخش گردن دراین نمایش سبب آسیب فوقانی گردنی می شود.ویژگی های بالینی:فلاسیدیته ،فقدان تنفس خودبخودی ،تنفس مشکل درانواع خفیف ،نیازبه تهویه مکانیکی درهمه موارد،کاهش سطح هوشیاری بعلت ادم ساقه مغز،فقدان رفلکس تاندونی درابتداوتشدیدآن درآینده،اتساع مثانه ،مرگ بعلت سپسیس یانارسائی تنفسی درهفته اول.تشخیص: MRIوانجام EMG به تشخیص کمک می کند.
اختلالات واحد حرکتی
ارزیابی اختلالات واحد حرکتی :انتخاب تستهای آزمایشگاهی درهیپوتونی مغزی بسته به بیماری فرق می کند(جدول).درحالیکه دراختلالات واحد حرکتی ، اینگونه نیست.مجموعه ای ازتست هامی تواندبه تعیین محل آناتومیک وعلت اختلال کمک کند.
| جدول:ارزیابی اختلالات واحد حرکتی |
|
· تست DNA · تست ادروفونیوم کلراید(تست تنسیلون ) · الکترودیاگنوز o الکترومیوگرافی o بررسی هدایت عصب o تحریک مکرر · بیوپسی عضله · بیوپسی عصب · کراتین کینازسرم |
انواع اختلالات واحد حرکتی
آتروفی های عضلانی نخاعی( SMAs):دراینگروه ازاختلال ژنتیک ، سلولهای شاخ قدام درنخاع وهسته های موتورساقه مغز بطورپیشرونده ای ازبین می روند.احتمالامکانیسم آن ،تسریع درمرگ سلولی است.شروع ضعف ازبدوتولدتابلوغ است.انواعی که درنوزادی تظاهرمی کنند،سبب ضعف ژنرالیزه وهیپوتونی می شوند. SMA نوزادی یکی ازشایعترین اختلالات واحد حرکتی است که سبب هیپوتونی نوزادی میشود.
SMA: شامل موارداست:
- نوع کشنده وشدید (SMA type 0) : که دردوره پری ناتال کشنده است.
- نوع شدید انفانتیل ( SMAI)[ Werdnig-Hoffmann disease] که همیشه قبل از6 ماهگی تظاهرمی کند،
- نوع بینابینی یا انفانتیل تاخیری (SMAII)، و
- نوع مزمن یا جونایل ( SMAIII)[ Kugelberg-Welander disease] .
- گونه ای بنام Fazio-Londe disease : که فلج پیشرونده بولبارناشی ازدژنراسیون نرون حرکتی بیشتردرساقه مغزمی باشد.
سایرانواع درجدول آمده است.
طبقه بندی بالینی آتروفی عضلانی نخاعی (SMA)
| SMA TYPE | AGE OF ONSET | HIGHEST FUNCTION | NATURAL AGE OF DEATH |
| Type 1 (severe) | 0-6 mo | Never sits | <2 yr |
| Type 2 (intermediate) | 7-18 mo | Never stands | <2 yr |
| Type 3 (mild) | >18 mo | Stands and walks | Adult |
| Type 4 (adult) | Second or third decade | Walks during adult years | Adult |
واریان های آتروفی عضلانی نخاعی :بیماری شدید یاپیشرونده سلول شاخ قدام که ارتباطی با SMN ندارند.
| واریان | ویژگی های اصلی |
| SMA with respiratory distress type 1 (SMARD1) | هیپوتونی خفیف ، گریه ضعیف ، کنتراکچوردیستال ازابتدا، دیسترس تنفسی ناشی ازفلج دیافراگم 6-1 ماهگی ،ضعف پیشرونده دیستال
اتوزوم مغلوب |
| Pontocerebellar hypoplasia type 1 | آرتروگریپوز،هیپوتونی ،ضعف ،نقص بولبارزودرس ،بعدها میکروسفالی ،نقائص اکسترااوکولار، نقائص شناختی ،هیپوپلازی پونتوسربلار
احتمالا اتوزوم مغلوب |
| X-linked infantile SMA with bone fractures | آرتروگریپوز، هیپوتونی ،ضعف ،شکستگی مادرزادی استخوان ،نارسائی تنفسی ،سیرکشنده نظیر SMAI
اکثرا وابسته به X ، برخی موارداتوزوم مغلوب |
| Congenital SMA with predominant lower limb involvement | آرتروگریپوز، هیپوتونی ،ضعف ،بخصوص ضعف زودرس دیستال اندام تحتانی
پیشرونده نیست ولی ناتوانی شدیدایجادمی کند. اتوزوم غالب یا اسپورادیک |
ژن نرون بقاء حرکتی(SMN1) ، عامل بیماری است .افرادنرمال حامل دوژن SMN1 و SMN2 می باشند.ویژگیهای بالینی نوع شدید دراینجامطرح می شود:سن شروع ازبدوتولدتا6 ماهگی است.کاهش حرکات جنینی دررحم،ضعف کلی بدن که عضلات پروگزیمال رابیشترازدیستال گرفتارمی کند،هیپوتونی ،و آرفلکسی .بعلت نرمال بودن عضلات صورت وحرکات اوکلوموتور،اکثراین نوزادان درابتداتنفس نرمالی داشته وظاهرهوشیاری دارند.برخی نیزدچارتنفس پارادوکس ( بعلت گرفتاری قفسه سینه وسالم ماندن دیافراگم ) می شوند.علیرغم هیپوتونی داخل رحمی،آرتروگریپوزدیده نمی شود.ضعف ، پیشرونده است .زبان دچارآتروفی وفاسیکولاسیون است هرچندکه تشخیص آن درنوزادمشکل می باشد.باازبین رفتن رفلکس گگ ، تغذیه مشکل می شودومرگ بعلت آسپیراسیون اتفاق می افتد.هرگاه علائم دربدوتولدباشند، مرگ معمولا در6 ماهگی رخ میدهد.تشخیص : باتست ژنتیک مولکولی است.بنابراین نیازی به بیوپسی عضله نمی باشد.امادربیوپسی گروه فیبرهای کوچک مجاورگروه فیبرهای درشت یانرمال دیده می شود.
| Werdnig-Hoffmann disease |  |
| آتروفی عضلانی نخاعی :
سمت چپ : SMAIII سمت راست : SMAII
|
 |
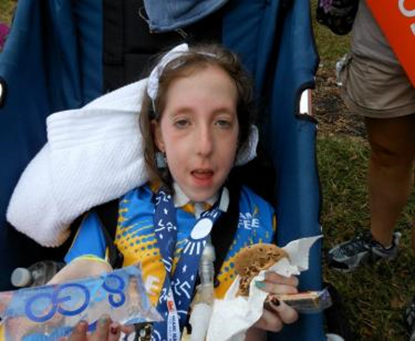
| تصویرچپ : دختر5 ماهه مبتلا به SMA نوع 1
تصویرمیانی :پسربچه مبتلا به SMA نوع 2 ( بابریس کمکی ) تصویرراست :پسرمبتلا به SMA نوع 3
|
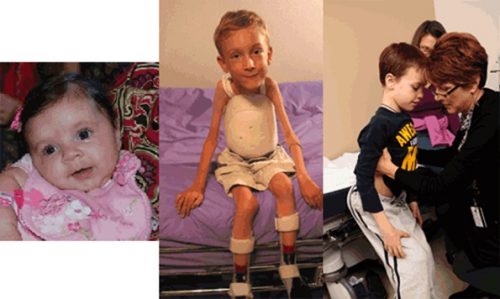 |
| Kugelberg-Welander disease |  |
 |
| A. شیرخوار2 روزه مبتلا به Congenital cervical spinal atrophy
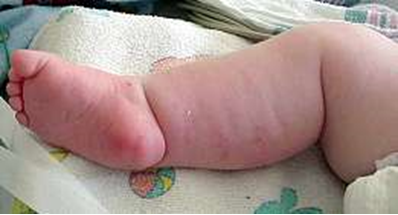
B. فلج شل محدود به اندام فوقانی توام با کنتراکچور فلکسیون مادرزادی C. لاغری وآتروفی عضلات انترنسیک دست توام با کنتراکچورفلکسیون انگشتان توام با تکامل ناقص شیارعرضی کف دستی |
آتروفی عضلانی نخاعی گردنی مادرزادی (Congenital Cervical Spinal Muscular Atrophy):یک اختلال نادراسپورادیک باضعف شدیدوتحلیل عضلانی محدودبه بازوهاست.پیشرفت ضعف بعدازتولد، دیده نمی شود.
آرتروگریپوزنروژنیک:انتقال بیماری بصورت اتوزوم مغلوب وگاهی وابسته به X می باشد.مرحله فعال بیماری درون رحم اتفاق می افتد.نوزادن بشدت مبتلادچارعلائم تنفسی وتغذیه ای بوده وبرخی بعلت آسپیراسیون فوت می شوند.دربرخی نیزباگذشت زمان، بهبودی دیده می شود.کنتراکچوردرمفاصل پروگزیمال ودیستال ومیکروگناتی ،کام بلند وآنومالی صورت وجوددارد.تشخیص :باآرتروگریپوز،غلظت نرمال CK ویافته های نروپاتیک در EMG می باشد.
کمبودسیتوکرام – سی اکسیداز:ازلحاظ بالینی یک گروه ناهمگون است وازمیوپاتی ایزوله تا کاردیوانسفالومیوپاتی کشنده نوزادی متفاوت است.در EMG، امواج مثبت تیزوپتانسیل فیبریلاسیون همراه باسرعت هدایت عصبی نرمال دیده می شود.
پلی نروپاتی ها:درکودکان نادربوده ودرطی نوزادی نادرترهستند.درجدول زیر فهرست پلی نروپاتی هائی که درنوزادی شروع می شوند ، آمده است . این اختلالات در2 گروهند : گروهی که میلین رامبتلا می سازند (دمیلینزاسیون ) یا آکسون را (آکسونال) .فقط درنروپاتی هیپومیلیناسیون مادرزادی اولین تظاهربیماری بصورت هیپوتونی نوزادی است.سایرانواع بصورت اختلال پیشرونده راه رفتن یا عقب افتادگی سایکوموتورتظاهرمی کنند.
| جدول :پلی نروپاتی های باشروع درنوزادی | |
| آکسونال
o دیس اتونومی فامیلیال o نروپاتی حسی حرکتی ارثی نوع 2 o ایدیوپاتیک همراه باانسفالوپاتی o دژنراسیون نرونی انفانتیل o انسفالوپاتی نکروزان تحت حاد |
دمیلیناسیون
o پلی نروپاتی حاددمیلیناسیون التهابی (سندروم گیلن باره) o پلی نروپاتی مزمن دمیلیناسیون التهابی o نروپاتی هیپومیلیناسیون مادرزادی o لکودیستروفی سلول گلوبوئید o نروپاتی حسی حرکتی ارثی نوع 1 o نروپاتی حسی حرکتی ارثی نوع 3 o لکودیستروفی متاکروماتیک |
نروپاتی هیپومیلیناسیون مادرزادی(neuropathy hypomyelinating congenital):شامل چندین اختلال باویژگیهای بالینی وپاتولوژیک شبیه هم است.بروزبیماری اسپورادیک وگاهی اتوزوم مغلوب است.ویژگیهای بالینی:نظیرآتروفی عضلانی نخاعی حادنوزادی است .آرتروگریپوزوجوددارد.ضعف فلاسید پیشرونده وآتروفی عضلات اسکلتی ، فلج بولبار (باعدم گرفتاری حرکات اکسترااوکولار) وآرفلکسی دیده می شود.نارسائی تنفسی باعث مرگ نوزادی است.تشخیص:غلظت CK نرمال است ویافته های EMG بیانگرعصب زدائی بوده وسرعت هدایت عصب حرکتی معمولا کمتراز10m/sec می باشد.غلظت پروتئین درمایع نخاعی بشدت بالاست.درمان:کورتون؟
|
Congenital Hypomyelinating Neuropathy |
 |
|||
|
Giant Axonal Neuropathy |
 |
 |
 |
|
|
تصویرچپ : Distal Hereditary Motor neuropathy تصویرراست : Hereditary Motor Sensory neuropathy
|
 |
 |
||
|
Hereditary sensory and autonomic neuropathy (Riley-Day Syndrome) به ویژگی های صورت بیمارمبتلا به HSAN درطول زمان ، بخصوص پهن شدن لب فوقانی ، دقت کنید.درسن 10 سالگی ،برجستگی فک تحتانی بخوبی بارزشده ودر19 سالگی ، تخریب خفیف پره بینی راست ، ناشی ازخودآزادی ناخواسته ، دیده می شود.
|
 |
|||
| Kyphoscoliosis_hereditary_sensory_autonomic_neuropathy_III | 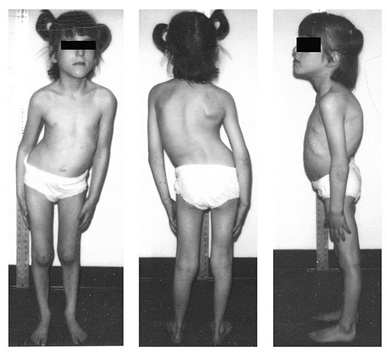 |
|||
اختلالات انتقال نروماسکولار
- بوتولیسم نوزادی
- سندرومهای میاستنیک مادرزادی
- میاستنی گذرای نوزادی
 |
 |
 |
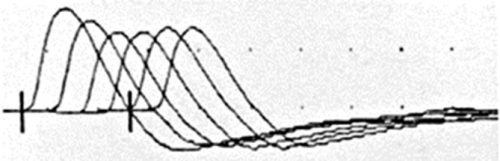
| کاهش موج درتحریک مکررعصب ، درمیاستنی گراویس | میاستنی گراویس : به پتوزچشم راست (تصویرچپ) وپتوزدوطرفه (تصویرراست) دقت کنید. | |
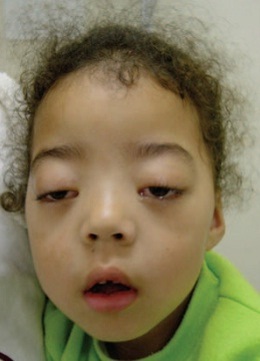
| میاستنی گراویس
به پتوزدوطرفه توجه کنید. |
 |
|
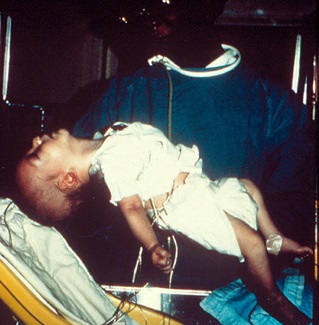
| بوتولیسم نوزادی : بیش از95% موارددرسنین 3 هفتگی تا6 ماهگی دیده می شود.
بوتولیسم ناشی ازتوکسین رهاشده ازکلستریدیوم بوتولینوم می باشد.نوع شیرخوارگی آن بیشترناشی ازمصرف موادغذائی آلوده همچون عسل می باشد.یافته هابصورت فلج شل متقارن ونزولی است که ابتداازعضلات اعصاب کرانیال شروع می شود. |
 |
|
میوپاتی های مادرزادی : تظاهراصلی بصورت هیپوتونی نوزادی و تشخیص بکمک بیوپسی است.یافته شایع هیستولوژیک بصورت تعدادزیادفیبرهای نوع 1 که کوچکترازنوع 2 هستند ، می باشد.
بیماری سنترال کور(Central Core Disease ):وراثت بصورت اتوزوم غالب ،وگاهی اتوزوم مغلوب واسپورادیک ، است.ویژگیهای بالینی:هیپوتونی خفیف پس ازتولدیاطی شیرخوارگی ، دررفتگی مادرزادی هیپ ،شروع ضعف باپیشرفت آرام پس ازسن 5 سالگی ،ضعف عضلانی درپروگزیمال بیشترازدیستال ودربازوهابیشترازپاهاست.رفلکس تاندونی عضلات ضعیف ،کاهش یافته یا هیچ است. حرکات اکسترااوکولار،حرکات صورت وبلعیدن نرمال است.بسیاری ازاین بیماران درمعرض خطرهیپرترمی بدخیم هستند.تشخیص:غلظت CK نرمال ویافته های EMGنیزنرمال است.البته EMG دراغلب مواردبیانگرفرایندهای میوپاتیک است.اساس تشخیص بابررسی هیستوپاتولوژی است که درآن میوفیبریلهای بشدت کنارهم که دستخوش دژنراسیون بوده ودرمرکزفیبرهای نوع 1 قراردارند، دیده می شود.
 |
 |
| central core disease
به پتوزدوطرفه و استرابیسم دقت کنید. |
central core disease |
| Central core disease
بیمار5 ساله مبتلابه سنترال کور، بعلت کنتراکچورشدیدهیپ وزانو فقط توانائی ایستادن دروضعیت فلکسیون به جلو (ازناحیه هیپ) وتکیه دست برزانو دارد.
|
 |
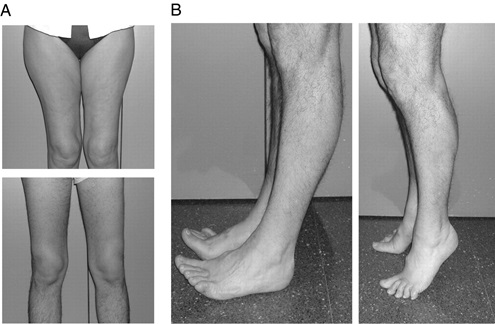
میوپاتی مادرزادی عدم تناسب نوع فیبر ( Congenital Fiber-Type Disproportion Myopathy): هردوجنس بطوریکسان گرفتارمی شوند.اکثرموارداسپورادیک است ، ولی انواع اتوزوم مغلوب ، غالب ووابسته به X نیزدارد.ویژگیهای بالینی:شدت ضعف بیماری ازهیپوتونی خفیف تانارسائی تنفسی متفاوت است.بسیاری دچارهیپوتونی داخل رحمی وبنابراین دررفتگی هیپ ، خصوصیات دیس مورفیک وکنتراکچورمفصلی هستند.عضلات پروگزیمال ضعیف ترازدیستال هستند.ضعف صورت ،کام بلند،پتوز،واختلال درحرکت چشم دیده می شود.درصورت ضعف آکسیال ،احتمال بروزکیفواسکولیوزدرکودکی وجوددارد.هوش نرمال است.تشخیص:غلظت CK نرمال یااندکی بالاست، EMGبیانگرفرایندهای نروپاتیک یامیوپاتیک ویاهردومی باشد.سرعت سیرعصب، نرمال می باشد.دربیوپسی ،هیپوتروفی وارجحیت فیبرنوع 1 دیده می شود.
 |
|
Congenital Fiber-Type Disproportion Myopathy A. آتروفی کوادریسپس که درتصویربالاوپائین قابل مشاهده است. B. ضعف پا و دورسی فلکسیون انگشتان پا دیده می شود، که البته هنگام ایستادن روی نوک انگشتان وجودندارد. |
بیماری مولتی – مینی کور(Multi-Minicore Disease):
میوپاتی میوتوبولار(سنترونوکلئار):
- میوپاتی میوتوبولارانفانتیل/شدید:
- میوپاتی میوتوبولاربینابینی / خفیف:
میوپاتی نمالین (رود)[ Nemaline (Rod) Myopathy]:وراثت بصورت اتوزوم غالب یامغلوب است.ویژگیهای بالینی:ضعف وهیپوتونی همراه باکاهش یافقدان رفلکس های تاندونی.ضعف عضلانی معمولا درناحیه صورت ،فلکسورهای گردنی،وعضلات پروگزیمال اندام شدیدتراست.تشخیص:غلظت CK معمولانرمال ویااندکی بالاست.بیوپسی اساس تشخیص می باشد.
|
Nemaline (Rod) Myopathy |
 |
|
|
Nemaline (Rod) Myopathy کنتراکچوردیستال همراه با ضعف عضلانی دراندام فوقانی وتحتانی . |
 |
 |
|
Nemaline (Rod) Myopathy |
 |
|
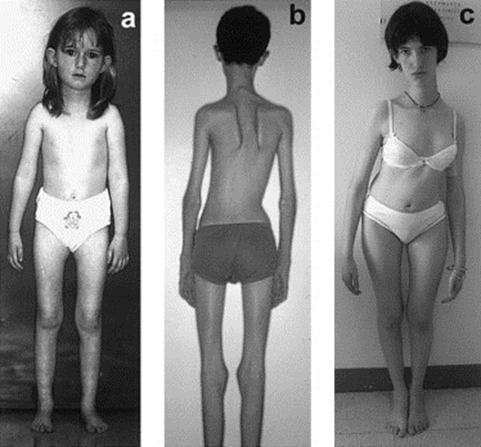
| بیماری مولتی مینی کور(Multiminicore )، یک اختلال اتوزوم مغلوب است که مشخصه آن وجودضایعات متعددباهسته کوتاه(مینی کور) دراکثرالیاف عضلانی است.
a) بیمار5.5 ساله باگردن باریک وفقدان توده عضله استرنوکلیدوماستوئید ،قفسه سینه مسطح و آتروفی متوسط دلتوئید ، آمیوتروفی سطح درونی ران ،ساق قلمی وپاهای مسطح. اسکولیوز، الگوی خاصی داردبگونه ای که بالوردوزدورسال (hollow back) وانحراف لترال تنه همراه است واغلب نیازمند آرترودزوسیع می باشد. b) بیمار15 ساله مبتلا به بیماری مولتی مینی کور c) بیمار17 ساله مبتلابه بیماری ، پس ازاصلاح جراحی برای اسکولیوز
|
 |
|
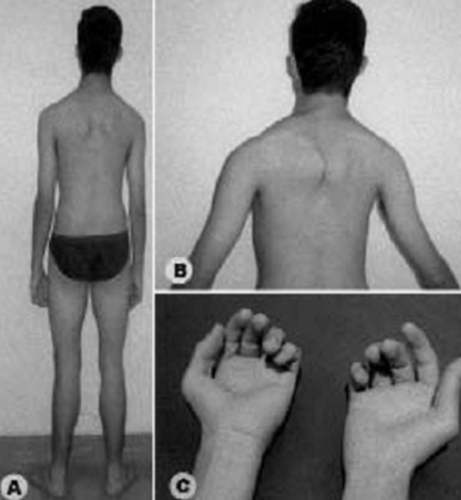
| بیماری مولتی مینی کور
به شبیه بال شدن(Winging) اسکاپولا وضعف کمربندشانه ای (a و b) وظاهرنرمال کمربندلگنی ودفورمیته مادرزادی دست ها (وناتوانی دراکستانسیون انگشتان دست ،c) توجه کنید. |
 |
|
دیستروفی های عضلانی
- دیستروفینوپاتی های مادرزادی:اغلب سبب ضعف دربدوتولدمی شوند.دراین موارد،دیستروفین بطورکامل وجودندارد.
- دیستروفی عضلانی مادرزادی: CMDگروهی ازمیوپاتی هاست که باهیپوتونی درتولدیااندکی بعد، کنتراکچورمتعددمفاصل ازاوایل حیات ،ضعف وآتروفی منتشرعضلات مشخص می شوند.وراثت بصورت اتوزوم مغلوب است. CMD رابه دوگروه سندرومیک (گرفتاری عضله ومغز) وغیرسندرومیک (بیماری عضلانی بدون گرفتاری مغزی) تفکیک می کنند.برخی نیز CMD رابراساس وجودمروزین (Merosin) تقسیم می کنند.
انواع CMD :
- Bethlem myopathy
- CMD with rigid spine syndrome
- Fukuyama CMD (FCMD)
- Integrin-deficient CMD
- Merosin-deficient CMD
- Muscle-eye-brain disease (MEB)
- Ullrich CMD
- Walker-Warburg syndrome(WWS)
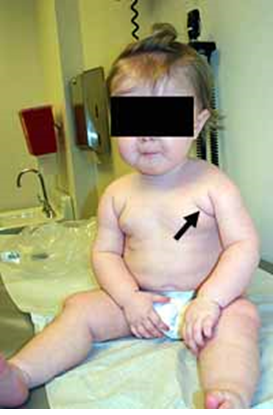
| Congenital muscular dystrophy: Merosin (laminin α2-chain) deficient
به چین های پکتورال (پیکان) همراه باضعف شدید شانه توجه کنید.
|
 |
||
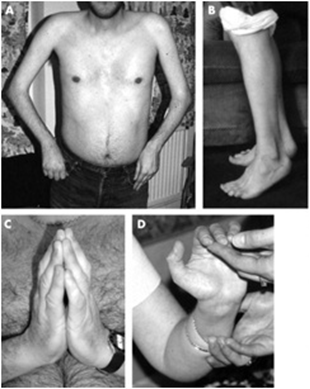
| میوپاتی Bethlem
تصویرچپ : به کنتراکچورفلکسیون انگشت دست ، کنتراکچورآرنج و تاندون آشیل( راه رفتن روی نوک انگشتان ) توجه کنید. تصویرراست : کلوئیدازیافته های این میوپاتی است.
|
 |
 |
|
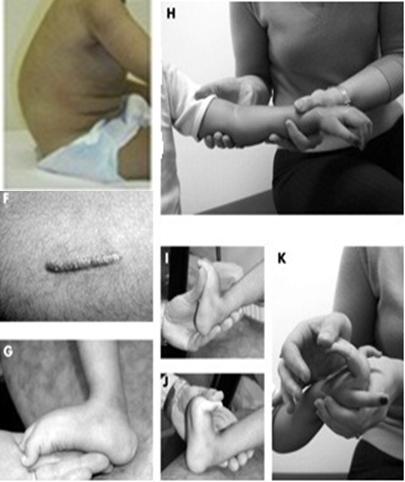
| Ullrich CMD
به هیپرفلکسیون انگشتان دست ،کالکانئوس برجسته ، کیفوزوکلوئیدتوجه کنید. |
 |
||
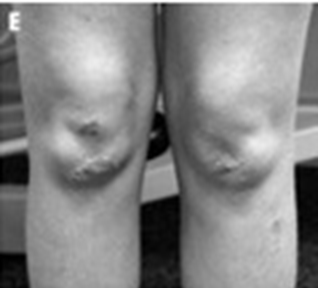
| انحراف جانبی ستون مهره ها درمیوپاتی مولتی کور ، ودیستروفی عضلانی رژید1(Rigid spine muscular dystrophy 1 ) (تصویرراست) ،رژیدیته زودرس دردیستروفی عضلانی مادرزادی(تصویرچپ)
|
 |
 |
|
دیستروفی عضلانی مادرزادی سندرومیک:دست کم درسه اختلال زیر، CMD توام باگرفتاری سیستم عصبی مرکزی می باشد.ویژگی اصلی ،آشفتگی درمهاجرت سلولی بسوی کورتکس مغزاست که سبب پلی میکروژیریا،لیزنسفالی ،وهتروتوپی می شود.سایریافته ها:الحاق لوب های فرونتال، آتروفی عصب اپتیک،هیپوپلازی مسیرپیرامیدال،کاهش تعدادسلولهای شاخ قدام
- دیستروفی میوتونیک مادرزادی:وراثت بصورت اتوزوم غالب است.علائم بیماری معمولادردهه دوم ظاهرمی شود.ویژگی اصلی دربارداری ،کاهش حرکات جنین و پلی هیدرامنیوس می باشد.نیمی پره ماچوربدنیامی آیند.ویژگیهای بالینی :دیپلژی صورت(بگونه ایکه لب فوقانی بصورت عدد8 فارسی است)،هیپوتونی ژنرالیزه ،دفورمیته مفاصل ازکلاب فوت دوطرفه تا آرتروگریپوزژنرالیزه،واختلال گوارشی ،ضعف عضلانی بیشترپروگزیمال است تادیستال.رفلکس های تاندونی معمولا درعضلات مبتلاوجودندارند.تشخیص:بامعاینه مادرمی توان تشخیص راشروع کرد.مادران بسیاری ازعلائم راداشته ودر EMG، میوتونی دارند.
- سایرسندروم های میوتونیک : اکثربیماران مبتلا به میوتونی دچاردیستروفی میوتونیک هستند، ولی میوتونی خاص اینگروه ازاختلالات نبوده ودرسایرمواردنیزدیده می شود.خلاصه اینگروه درجدول آمده است.
کانالوپاتی ها واختلالات مرتبط
|
کانالوپاتی های کلراید · میوتونیا کونژنیتا (Myotonia Congenita ) o بیماری تامسن (Thomsen) :میوتونی ،اتوزوم غالب o بیماری بکر(Becker) :میوتونی وضعف ، اتوزوم مغلوب کانالوپاتی های سدیم · پارامیوتونی کونژنیتا : پارامیوتونی ، اتوزوم غالب · فلج پریودیک هیپرکالمیک : فلج متناوب توام با میوتونی و پارامیوتونی ، اتوزوم غالب · فلج پریودیک هیپوکالمیک :فلج متناوب ، اتوزوم غالب میوتونی تشدیدیابنده باپتاسیم · میوتونیا فلاکچوآن (fluctuans ) : میوتونی ،اتوزوم غالب · میوتونیا پرماننز(permanens) : میوتونی ، اتوزوم غالب · میوتونی پاسخ دهنده به استازولامید :میوتونی ،اتوزوم غالب کانالوپاتی کلسیم · فلج پریودیک هیپوکالمیک : فلج متناوب ،اتوزوم غالب · میوتونی کندرودیستروفیک ( سندروم Schwartz-Jampel) · بیماری عضلانی ریپلینگ (Rippling) : برجستگی عضله ، سفتی ، اتوزوم غالب · سندروم آندرسون (Anderson) : فلج متناوب ،آریتمی قلبی ، صورت خاص · بیماری برودی (Brody ) :ریلاکسشن تاخیری ، فقدان میوتونی الکترومیوگرافیک ، اتوزوم مغلوب · هیپرترمی بدخیم :ریلاکسشن تاخیری ناشی هوشبری ، اتوزوم غالب
|
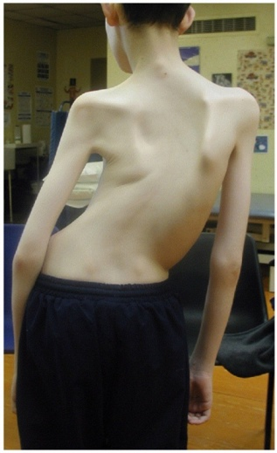
| دیستروفی دوشن :
تصویرچپ : هیپرتروفی کاذب دربیماربزرگسال تصویرراست:به الگوی عمده میوپاتیک همراه باآتروفی پروگزیمال ، تشدید لوردوزکمری ، هیپرتروفی ورتراکسیون تاندون آشیل الگوی هیپوتروفی شدیدومنتشرواسکاپولای بالدارتوجه کنید. |
 |
 |
|
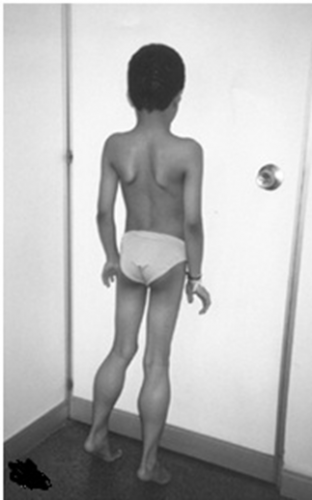
| Merosin-negative CMD
به ضعف ودفورمیته صورت توجه کنید. |
 |
||
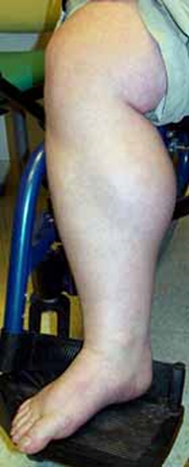
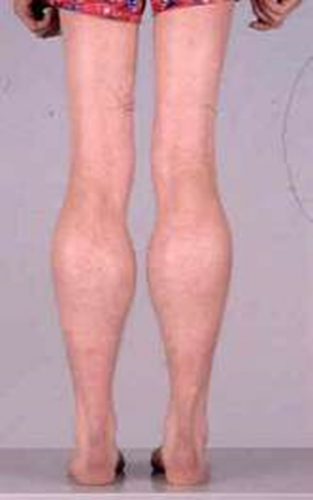
| Becker muscular dystrophy
به راه رفتن روی نوک انگشتان پا (تصویرچپ) وبزرگی عضله پشت ساق پا توجه کنید. |
 |
 |
|
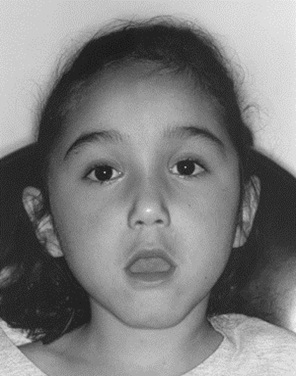
| myotonic muscular dystrophy
ضعف عضلات صورت ،لب فوقانی بشکل 8 فارسی ،فقدان توده عضلانی درحفره تمپورال ازویژگی های دسیتروفی عضلانی میوتونیک است. |
 |
||
| Facioscapulohumeral Muscular Dystrophy | 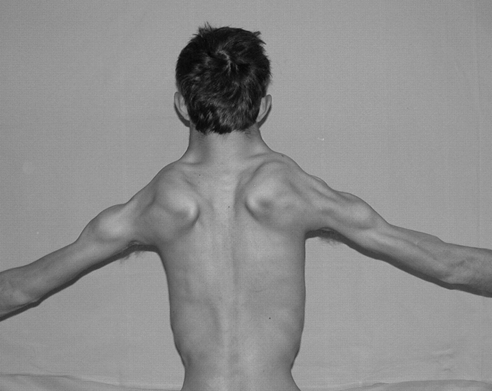 |
||
میوپاتی های متابولیک
کمبوداسیدمالتاز، بیماری پومپه(Acid maltase deficiency)
 |
 |
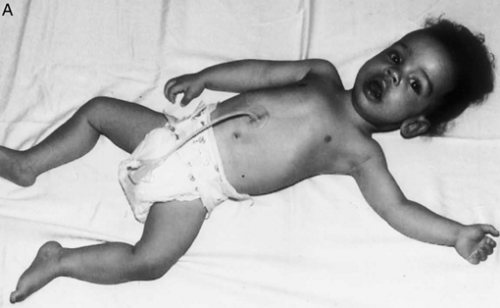
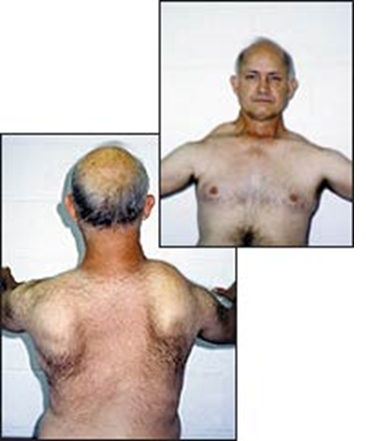
| کمبوداسیدمالتاز(بیماری پومپه ) : به هیپوتونی جنرالیزه دقت کنید. |
Acid maltase deficiency
|
 |
 |
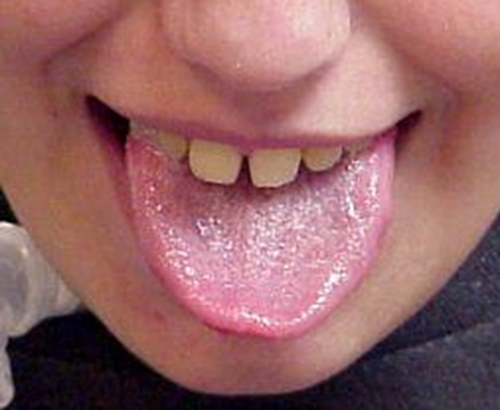
| بیماری پومپه : به بزرگی خفیف زبان دقت کنید. | Acid maltase deficiency |


{kind=link}
{kind=link}
{kind=link}