

پلاژیوسفالی تغییر شکلی
اسفند ۴، ۱۴۰۲
دکتر قمر حسینی الهاشمی
اسفند ۵، ۱۴۰۲
سندروم صورت خندان
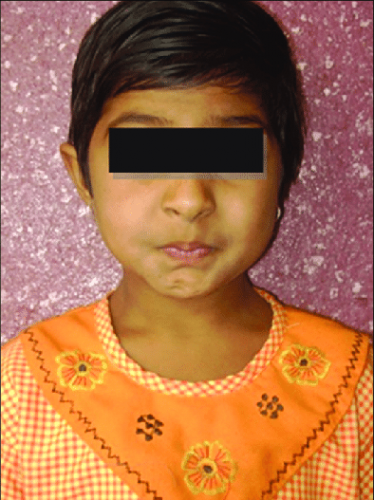
سندرم Freeman-Sheldon یک اختلال است که ابتدا صورت ، دست ها و پاها را تحت تاثیر قرار میدهد. افراد مبتلا به این بیماری دارای چهره ای متفاوت میباشند که از جمله میتوان به دهان کوچک ( میکروستومیا) همراه با لب های جمع شده اشاره کرد که به صورت فرد ، حالتی شبیه به حالت سوت زدن میدهد. به همین دلیل ، گاهی این بیماری( سندرم صورت سوت زننده ) نامیده میشود. افراد مبتلا به این سندرم ممکن است بعلاوه دارای پیشانی و خط ابروی برجسته ، فرورفتگی میانه صورت ، بینی کوتاه ، فاصله زیاد بین بینی و دهان ، چین های عمیق در پوست بین بینی و لب ها ، گونه های پر و گودی چانه به شکل حرف H یا V ، باشند. افراد مبتلا ممکن است دارای یک سری اختلالات باشند که چشم ها را تحت تاثیر قرار میدهند. این اختلالات ممکن است شامل چشم هایی با فاصله زیاد ( هایپرتلوریسم ) ، چشم هایی گود ، چشمهایی که گوشه های خارجی آن ها به سمت پایین تمایل دارند ، باریک بودن شکاف چشم ( بلفاروفیموزیس ) ، افتادگی پلک چشم ( پتوز ) و چشم هایی که در یک جهت نگاه نمیکنند ( استرابیسم ) ، باشند.
دیگر ویژگیهای چهره ای که ممکن است در این سندرم ظاهر شوند شامل زبان و فک غیرطبیعی کوچک ( میکروگلوسیا و میکروگناتیا ) و کمان بلند در سقف دهان میباشند. افراد مبتلا به این بیماری ممکن است دارای اختلال در بلع ( دیس فاژی ) ، ناتوانی در وزن گرفتن و رشد قابل انتظار ( نارسایی رشدی ) و عارضه های تنفسی گاها مخاطره آمیز ، باشند. مشکلات گفتاری نیز در این بیماری شایع هستند. برخی افراد مبتلا ناشنوا نیز میباشند. سندرم Freeman-Sheldon از طریق بدشکلی های مفصلی که حرکت را محدود میکنند نیز شناسایی میشود. افراد مبتلا به این بیماری معمولا دارای انقباض های متعددی در دست ها و پاهای خود در زمان تولد میباشند ( distal arthrogryposis ). این انقباضات منجر به خمیدگی دائمی انگشت های دست و پا ( کمپتوداکتیلی ) ، یک بدشکلی در دست که در آن تمام انگشت ها به سمت بیرونی انگشت پنجم ( انگشت کوچک ) خم شده اند ( انحراف اولنار ) و پاهایی که به سمت داخل و پایین خم شده اند ( پای چماقی ) میشوند. افراد مبتلا ممکن است دارای انحنای جانبی ستون فقرات ( اسکولیوز ) نیز باشند.
 |
 |
 |
افراد مبتلا به این سندرم دارای افزایش ریسک ایجاد یک واکنش شدید به داروهای خاصی که حین عمل و دیگر فرآیندهای تهاجمی مورد استفاده قرار میگیرند نیز میباشند. این واکنش هایپرترمی بدخیم نامیده میشود. هایپرترمی بدخیم در پاسخ به برخی گازهای بی حسی که برای مسدود کردن حس درد مورد استفاده قرار میگیرند ، رخ میدهد. یک نوع ویژه شل کننده عضلانی نیز ممکن است واکنش را راه اندازی کند. اگر این داروها مصرف شوند ، افراد در معرض خطر هایپرترمی بدخیم ممکن است سختی عضلانی ، تجزیه رشته های عضلانی ( رابدومیولیز ) ، تب بالا ، افزایش میزان اسید در خون و دیگر بافت ها ( اسیدوز ) و تندی ضربان قلب را تجربه کنند. عوارض هایپرترمی بدخیم ممکن است تهدید کننده زندگی فرد باشند مگر اینکه فورا درمان شوند.
فراوانی
سندرم Freeman-Sheldon یک اختلال نادر است ؛ شیوع دقیق آن مشخص نیست.
علل بیماری
سندرم Freeman-Sheldon ممکن است در اثر جهش های ژن MYH3 ایجاد شود. ژن MYH3 دستورالعمل هایی برای ساخت پروتئینی به نام زنجیره سنگین میوزین عضله اسکلتی جنینی شماره 3 فراهم میکند. این پروتئین به گروهی از پروتئین ها به نام میوزین ها تعلق دارد که در حرکت سلولی و انتقال مواد درون و بین سلول ها مشارکت میکنند. میوزین و پروتئین دیگری به نام اکتین اجزای اولیه رشته های عضلانی بوده و برای انقباض عضلات ضروری هستند. زنجیره سنگین میوزین عضله اسکلتی جنینی شماره 3 بخشی از یک کمپلکس پروتئینی میوزین را تشکیل میدهد که قبل از تولد فعال بوده و برای تکوین طبیعی عضلات ضروری است. جهش های ژن MYH3 که باعث ایجاد سندرم Freeman-Sheldon میشوند احتمالا عملکرد پروتئین زنجیره سنگین میوزین عضله اسکلتی جنینی شماره 3 را مختل کرده باعث کاهش توانایی انقباض سلول های عضلانی جنینی میشوند. این نقص در انقباض عضلانی ممکن است با تکوین عضلانی در جنین مداخله کرده و منجر به انقباضات و دیگر آنومالی های عضلانی و اسکلتی مرتبط با سندرم Freeman-Sheldon شود. اینکه چگونه جهش های ژن MYH3 ممکن است باعث دیگر ویژگیهای این اختلال شوند مبهم است. برخی افراد مبتلا به سندرم Freeman-Sheldon دارای جهش در ژن MYH3 نمیباشند. در این افراد ، علت بیماری مشخص نیست.
الگوی توارث
سندرم Freeman-Sheldon میتواند الگوهای توارث متفاوتی داشته باشد. در برخی موارد ، بیماری با الگوی توارث اتوزومال غالب به ارث میرسد ، به این معنی که یک کپی از ژن تغییریافته در هر سلول برای ایجاد بیماری کافی است. این بیماری میتواند الگوی توارث اتوزومال مغلوب نیز داشته باشد ، به این معنی که هر دو کپی ژن در هر سلول حاوی جهش میباشند. والدین فردی با بیماری اتوزومی مغلوب هر کدام یک کپی از ژن جهش یافته را حمل میکنند ، اما معمولا علائم و نشانه های بیماری را نشان نمیدهند. در برخی موارد ، الگوی توارث نامشخص است.


{kind=link}
{kind=link}
{kind=link}